概述
染色體異常遺傳病(簡稱“染色體病”):特點是染色體的數目異常和形态結構畸變,可以發于每一條染色體上。綜合許多國家的資料,大約有15%的妊娠發生流産,而其中一半為染色體異常所緻,即約為5%-8%的胚胎有染色體異常。不過在出生前,90%以上已有自然流産或死産。流産愈早,有染色體異常的頻率愈高。
其分類有:常染色體異常、性染色體異常
常染色體異常
(一)三體綜合征
1.先天愚型
先天愚型是最重要的染色體疾病。英國醫生Langdon Down首先描述,故稱為Down綜合征(Down sydrome)。
1959年,法國細胞遺傳學家Lejeune證實此病的病因是多了一個小的G組染色體(後來确定為21号),故此病又稱為21三體綜合征。Lejeune的發現開創了醫學遺傳學的一個重要分支――臨床細胞遺傳學。
(1)發病率:新生兒中21三體綜合征的發病率約為1/800或1.25%,但男性患兒多于女性。母親年齡是影響發病率的重要因素。根據外國資料,如果一般人出生時的母親年齡平均28.2歲,則本病患兒母親平均年齡為34.4歲,如母齡20歲後為1:2000,35歲後為1:300,40歲後為1:100,45歲後升至1:50。
(2)臨床表現:先天愚型患兒出生時體重和身長偏低,肌張力低下,突出的是顱面部畸形,頭顱小而圓,枕部扁平,臉圓而扁平,鼻扁平,臉裂細且上外傾斜,眼距過寬,内眦贅皮明顯,常有斜視,虹膜時有白斑點,常見晶狀體混濁,嘴小唇厚,舌大外伸(伸舌樣癡呆之名由此而來),耳小,耳位低,耳廓畸形,頸背部短而寬,有多餘的皮膚,由于軟骨發育差,患者四肢較短,手寬而肥,通貫掌,指短,第5指常内彎,短小或缺小指中節,皮紋也有一定的特點(參閱第十章),腹肌張力低下而膨脹,故常有腹直肌分離或臍疝,約1/2以上的患者有先天性心髒病,主要是室間隔缺損、房室道連通、和運脈導管未閉,消化道的畸形如十二指腸的狹窄、巨結腸、直腸脫垂及肛門閉鎖等也偶爾可見。在男性常有隐睾,睾丸有生精過程,但精子常減少,性欲下降,尚未見有生育者。女性患者通常無月經,但有少數能妊娠和生育。精神發育遲滞或智力低下(mental retardation,MR)是本病最突出最嚴重的表現,但其程度在各患者不完全相同,智商通常在25-50之間,高于50的很少。行為、動作傾向于定型化,抽象思維能力受損最大。
(3)實驗室檢查:過氧化物岐化酶(SOD-1)活性可增高50%,該酶基因定位21q22,即具有基因劑量效應。此外,患者對阿托品特别敏感,患者乙酰膽堿缺陷或許可以解釋智力低下、應變力差、便秘等症狀,而免疫功能失調,如淋巴細胞和丙球蛋白減少則是患兒易感染的原因。
(4)核型:核型可分為三型,各型的比例是:典型的(遊離型),即47,+21占95%;嵌合型即46/47,+21
占1%-2%;易位型占3%-4%。遊離型全身體細胞均多一條21号染色體,臨床症狀典型而且顯著。由于嵌合型通常具有兩個細胞系,其症狀表現取決于異常細胞所占的比例,故差異很大,但一般較典型者為輕。如果三體細胞很少,則表現與正常人無異。易位型的核型有多種,最常見的是Dq21q,占全部易位型的54.2%,其次是21qGq,占40.9%,其它易位型5%。一般說來,易位型的症狀比典型的要輕些,在Dq21q中,最常見的是14q21q,占Dq21q的58.5%,其次為13q21q,占22%,而15q21q占19.5%。21qGq易位中,21q21q占83.3%,而21q22q僅占16.6%。無論體積易位,患者雖然隻有46條體,但因一條21号易位到了另一條D組或G組染色體上,加上正常的兩條21号,仍多了一條額外的21号長臂,而決定本病的關鍵區帶為21号長臂,故臨床上仍表現出21三體的症狀。
(6)遺傳學:典型的21三體幾乎都是新發生(denovo)的,與父母的核型無關,經是減數分裂時不分離的結果。不分主離常發生在母方生殖細胞,約占病例數的95%,另5%見于父方,而且主要發生在第一次減數分裂。典型的21三體隻有極少一部分是遺傳的,即母親是本病患者。此外,不能排除某些表型正常的母親實際是21三體細胞較少的嵌合體,因而她們的子女有可能獲得額外的21号染色體。男性患者不能生育,沒有遺傳給下一代的問題。
嵌合型患者有兩個或兩個以上的細胞系。它們是合子後(post-zygotic)有絲分裂不分離的結果。如果第一次卵裂時發生不分離,就會産生47,+21和45,-21兩個細胞系。而後一種細胞是很維存活的。因此,導緻嵌合體的不分離多半發生在以後的某次有絲分裂,所有嵌合體内都有正常的細胞系。
易位型的21三體征患者細胞中有一條易位的染色體,後者通常由一條D組或G組染色體與21号染色體長臂通過着絲粒融合(羅氏易位)而成。Dq21q易位中,55%是新發生的,45%是由于雙親之一有平衡易位。21qGq易位幾乎全部(96%)是新發生的,由遺傳而來的僅占4%。
各種易位的遺傳後果不同。Dq21q平衡易位的攜帶者通過減數分裂可以形成6種配子,而受精後除不能發育者外,可以産生正常胎兒、易位型三體患兒和平衡易位攜帶者三種胎兒。因此,檢出平衡易位攜帶者的雙親具有重要意義。
21qGq易位中,21q22q和21q21q易位的遺傳學意義不完全相同。如果雙親之一為21q21平衡易位攜帶者,就沒有可能娩出表型正常的胎兒,因為他們隻能産生三體或單體的合子。21q22q易位的遺傳後果與Dq22q相似,隻是前者更多通過父親傳遞而後者多由母親傳遞得來。
(6)遺傳咨詢:各種類型再發風險不同。對遊離型的21三體性,再發風險率與年齡特異風險率相近(即35歲以下約為0.5%,35%歲以上約為1%)。然而,如雙親(通常是母親)之一嵌合體則風險會增加。總之,已産生一個21三體患兒,其再發風險0.5%,而染色體畸變的總風險為1.2%,即不排除再生産其它染色體異常的患兒。
易位型有大約1/2的病例是新發生的,另1/2是雙親的之一平衡易位引起的。前者複發風險很小,而平衡易位導緻的再發風險則可以根據經驗估計。理論上如雙親之一為平衡易位攜帶者,産出患兒風險為33.3%。但實際風險低得多,這與雙親哪一方為攜帶者有關。Dq21q易位攜帶者若是母親,生産患兒的風險為10%-15%;如為父親,則風險為5%或更小。21q21q易位的情況與之大體相同,但易位染色體由父方傳遞的百分比較D/G易位要多,風險率在10%以下。21q21q平衡易位攜帶者的後代100%均是三體征患兒,攜帶者不宜生育。
(7)預後:患者平均壽命隻有16.2歲。50%的患兒在5歲以前死亡。隻有8%的患者超過40歲,2.6%超過50歲。根據四川省的資料,人群中患病率僅約為再生時的1/10。
2.13三體綜合征
1960年Patau首先描述本病,故又稱為Patau綜合征。新生兒中的發病率約為1:25000,女性明顯多于男性。
(1)臨床表現:患兒的畸形和臨床表現要比21三體性嚴重得多。顱面的畸形包括小頭,前額、前腦發育缺陷,眼球小,常有虹膜缺損,鼻寬而扁平,2/3患兒有上唇裂,并常有腭裂,耳位低,耳廓畸形,颌小,其它常見多指(趾),手指相蓋疊,足跟向後突出及足掌中凸,形成所謂搖椅底足。男性常有陰囊畸形和隐睾,女性則有陰蒂肥大,雙陰道,雙角子宮等。腦和内髒的畸形非常普遍,如無嗅腦,心室或心房間隔缺損、動脈導管未閉,多囊腎、腎盂積水等,由于内耳螺旋器缺損造成耳聾。
智力發育障礙見于所有的患者,而且程度嚴重,存活較久的患兒還有癫痫樣發作,肌張功力低下等。
(2)細胞遺傳學及遺傳咨詢:80%的病例為遊離型13三體,核型為46,XX(或XY),+13,其餘的則為嵌合型或易位型。嵌合型一般症狀較輕,易位型通常以13和14号羅氏易位居多,患者有一條t(13q14q)易位染色體,核型為46,-14,+t(13q14q),其結果是多了一條13号長臂,當雙親之一是平衡易位攜帶者時,因為絕大多數異常胎兒均流産死亡,産出患兒的風險不超過5%或1%。如果雙親之一為13q13q易位攜帶者,由于隻能産生三體或單體的合子,流産率達100%。
(3)病因及預後:與13三體發和表關的因素所知甚少,母親高齡可能是原因之一,患兒母親的平均年齡為31.6歲,父親的平均年齡為34.6歲。此外,有資料表明,79%病例妊娠于寒冷季節(9-2月),45%的患兒在出生後一個月内死亡,90%在6個月内死亡,存活至3歲者少于5%,平均壽命為130天。
3.18三體綜合征
1960年Edward等首先描述,故又稱為Edward綜合征(Edward syndrome).18三體性導緻嚴重畸形,在出生後不久死亡。發病率約1:3500-8000新生兒。但在某些地區或季節明顯增高,達到1:450-800。患兒中女性:男性比為4:1。
(1)臨床表現:患兒出生時體重低,平均僅2243g,發育如早産兒,吸吮差,反應弱,頭面部和手足有嚴重畸形,頭長而枕部凸出,面圓,眼距寬,有内贅眦皮,眼球小,角膜混濁,鼻梁細長,嘴小,耳位低,耳廓畸形(動物樣耳),小颌(micrognathia),頸短,有多餘的皮膚,全身骨骼肌發育異常,胸骨短,骨盆狹窄,臍疝或腹股溝疝,腹直肌分離等。手的畸形非常典型:緊握拳,拇指橫蓋于其它指上,其它手指互相疊蓋,指甲發育不全,手指弓形紋過多,約1/3患者為通貫掌。下肢最突出的是“搖椅底足”,拇趾短,向背側屈起。外生殖器畸形比較常見的有隐睾或大陰唇和陰蒂發育不良等。95%的病例有先天性心髒病,如室間隔缺損、動脈導管未閉等,這是死亡的重要原因。腎畸形,腎盂積水也很常見。患兒智力有明顯缺陷,但因存活時間很短,多數難以測量。
(2)細胞遺傳學:80%患者核型為47,XY(或XX),+18;另10%患者為嵌合體,即為46,XY(或XX)/47,XY(或XX),+18;其餘為各種易位,主要是18号與D組染色體的易位。雙親是平衡易位攜帶者而導緻18三體征者很少。
(3)預後;患兒大多在2-3個月内死亡,平均存活71天,隻有極個别病人超過兒童期。嵌合型的存活期比較長。
4.其它染色體三體綜合征
比較重要的有8号、22号三體綜合征等。都伴有明顯的發育畸形和智力低下。還有一系列由易位引起染色體部分三體綜合征,其臨床症狀取決于額外染色體片段的性質和大小。染色體部分三體性可分為兩大類:一類有某一染色體片段的三體性(重複),同時又伴有其它染色體的異常(如缺失、易位),這一類部分三體性的表型比較複雜,常兼有重複和缺失片段的某些症狀;另一類為染色體的某一片段的單純重複或三體性,這在人類極為少見。
(二)單體及部分單體綜合征
整條常染色體的丢失通常是緻死的,因而極為罕見,但确有小染色體(如21号)完全丢失的報告。由于易位、環形成或缺失導緻的染色體部分單體則比較多見。現以5q-綜合征,即貓叫綜合征為介紹如下。
貓叫綜合征
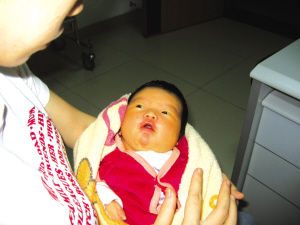
(5q-綜合征)為最見的缺失綜合征,其發病率估計為1:50000,女性多于男性。患嬰的哭叫聲非常似小貓的咪咪聲,故得名。患兒面部情似很機靈,但實則智力低下非常嚴重(智商常低于20),發育遲滞也很明顯。常見的臨床表現還有小頭、滿月臉、眼裂過寬、内眦贅皮、下颌小且後縮。約20%患者有先天性心髒病,主要是室間隔缺損和動脈導管未閉等。
患者的染色體缺失片段大小不一。症狀主要由5p15的缺失引起。畸變多數是新發生的。由染色體片段的單純缺失(包括中間缺失)的占80%,不平衡易位引起的占10%,環狀染色體或嵌全體則比較少見,由親代染色體重排導緻的5p-綜合征不多見。
性染色體異常
(一)性染色體數目異常綜合征
1.Klinefelter綜合征(Klinefelter syndrome)
又稱為先天性睾丸發育不全或原發小睾丸症。患者性染色體為XXY,即比正常男性多了一條X染色體,因之本亦常稱為XXY綜合征。
Klinefelter綜合征的發病率相當高,男性新生兒中達到1.2‰。根據白種人的資料,身高180cm的男性患病率為1/260,在精神病患者或刑事收容機構中為1/100,在因不育而就診者中約為1/20。臨床表現為睾丸小而質硬,曲細精管萎縮,呈玻璃樣變。由于無精子産生,故97%患者不育。患者男性第二性征發育差,有女性化表現,如無胡須,體毛少,陰毛分布如女性,陰莖龜頭小等,約25%的患者有乳房發戶。患者身材高。四肢長,一部分患者(約1/4)有智力低下,一些患者還有精神異常及患精神分裂症傾向。實驗室檢查可見雌激素增多,19-黃體酮增高,激素的失調與患者的女性化可能有關。
絕大多數患者的核型為47,XXY。大約有15%患者為兩個或更多細胞系的嵌合體,其中常見的為46,XY/47,XXY;46,XY/48,XXXY。額外的X是由于親代減數分裂時X染色體不分離的結果。
用睾丸酮治療可以收到一定的效果,它可促使第二性征發良并性病患者的心理狀态。
2.XYY綜合征
在男嬰中的發生率為1:900。XYY男性的表型的正常的,患者身材高大,常超過180cm,偶爾可見隐睾,睾丸發育不全并有精過程障礙和生育力下降,尿道下裂等,但大多數男性可以生育。XYY個體易于興奮,易感到欲望不滿足,厭學,自我克制力差,易産生攻擊性行為。XYY核型是父親精子形成過程中第二次減數分裂時發生Y染色體不分離的結果。
3.Turner綜合征
又稱為45,X或45,XO綜合征、女性先天性性腺發育不全或先天性卵巢發育不全綜合征。在新生女嬰中的發病率約為0.2‰-0.4‰,但在自發流産胚胎中Turner綜合征的發生率可高達7.5%.患者表型為女性,身材矮小,智力一般正常,但常低于其同胞,面呈三角形,常有睑下垂及内眦贅皮等,上颌突窄,下颌小且後縮,口角下旋呈鲨魚樣嘴,頸部的發際很低,可一直伸延到肩部,約50%患者有蹼頸,即多餘的翼狀皮膚,雙肩徑寬,胸寬平如盾,乳頭和乳腺發育差,兩乳頭距寬,肘外翻在本病十分典型,第四、第五掌骨短而内彎,并常有指甲發育不全。嬰兒期腳背有淋巴樣腫,十分特殊。泌尿生殖系統的異常主要是卵巢發育差(索狀性腺),無濾泡形成,子宮發育不全,常因原發性閉經來就診。由于卵巢功能低下患者的陰毛稀少,無腋毛,外生殖器幼稚。此外,大約有1/2患者有主動脈狹窄和馬蹄腎等畸形。
Turner綜合征的核型除典型的45,X(約占55%)外,還有各種嵌合型和結構異常的核型。最常見的是嵌合型46,XX/45,X和46,X,i(Xq)。一般說來,嵌合型的臨床表現較輕,而有Y染色體的嵌合型可表現出男性化的特征,身材矮小和其它Turner症狀主要是由X短臂單體性決定的,但卵巢發育不全與不育則更多與長臂單體性有關。
Turner綜合征的發病機理是雙親配子形成過程中的不分離,其中約75%的染色體丢失發生在父方,約10%的丢失發生在合子後早期卵裂時。
除少數患者由于嚴重畸形有新生兒期死亡之外,一般均能存活,隻是在青春期才被檢出。其智力發育障礙也較輕,應用激素在14歲以前開始治療可以促進第二性征和生殖器官的發育,月經來潮,心理狀态改變,但不能促進長高,個别患者可生育。
4.XXX和X女性
本病又稱為超雌(superfemale)。發病率約為0.8‰或1/2250。
多數具有三條X染色體的女性無論外形、性功能與生育力都是正常的,隻有少數患者有月經減少、繼發閉經或過早絕經等現象。大約有2/3病智力稍低,并有患精神病傾向。
除了47,XXX外,一些患者的核型為嵌合型,即47,XXX/46,XX。XXX患者的母親生育年齡平均約增高4歲。這表明不分離主要發生在母親一方。少數患者有4條甚至5條X染色體。一般來說,X染色體愈多,智力損害和發育畸形愈嚴重。
(二)性染色體的結構畸變
1.X染色體的結構異常
常見的X染色體結構異常有各種缺失、易位和等臂染色體。它們的臨床表現多樣,主要取決于涉及X染色體上的哪些區段異常,因為不同的區段載有的基因不同,缺失導緻的症狀也不同。Wyss等曾作一Turner綜合征的核型與症狀關系圖。有助于了解X染色體結構異常的表現。
(1)X短臂缺失(XXp-):Xp遠端缺失病人有諸如身材矮小等Turner綜合征特征,但性腺功能正常。Xp缺失如包括整個短臂,則患者既有Turner綜合征的體征,又有性腺發育不全。X染色體長臂等染色體[X,i,(Xq)]的臨床表現與此類似,因為也缺失了整個短臂。
(2)X長臂缺失(XXq-):缺失在q22以遠者,一般僅有性腺發育不全,原發閉經,不育,而無其它諸如身材矮小等Turner綜合征體征。缺失範圍較大,包括長臂近端者,有性腺發育不全外,一些患者還有其客觀存在體征。X染色體等臂染色體[Xi(p)]與此類似。Xq中間缺失累及q13-q26者性腺功能正常,但有其它體征,可見中段缺失與Turner體征出現有關。
通常部分缺失`形成環狀或等臂染色體的X均選擇性地失省事,從而保證有一條正常的X。
(3)易位:當X染色體與常染色體發生平衡易位時,由于基因平衡的保持,一般不會産生症狀,此時失活的正常的X染色體。但如平衡易位斷點在q12-q26時,有活性的X在該區被分為兩部分,就會導緻性腺發育異常。此外,如常染色體節段易位到X染色體産生不平衡易位時,多數産生雙着絲粒染色體,其表型取決于Xp或Xq上斷裂點的位置。
2.脆性X染色體綜合征
本世紀初一些學者注意到智力下患者中邏輯性多于女性。1943年Martin和Bell在一個家系兩代人中發現11名邏輯性患者和兩名輕度智力低下的女性,認為該家系智力低下是與X連鎖的,因此X連鎖智力低下又稱為Martin-Bell綜合征。
1969年Lubs首先在邏輯性智力低下患者及其女性親屬中發現了長臂具有“随體和呈細絲狀次缢痕”的X染色體。後來,Sortherland證明細絲位位于X染色體長臂2區7帶(Xq27)。它在低葉酸培養條件下表達,并提出了脆性部位(franile site)的概念。現今人們把在Xq27處有脆性部位的X染色體稱為X染色體(fragile X,fra X),而它所導緻的疾病稱為脆性X染色體綜合征。
(1)發病率:本病在邏輯性中的發病率為1/1000~1/1500,僅次于先天愚型。在所有邏輯性智力低下患者約10%~20~為本病所引起。
(2)臨床表現:主要表現為中度到重度的智力低下,其它常見的特征尚有身長和體重超過正常兒,發育快,前額突出,面中部發育不全,下颌大而前突,大耳,高腭弓,唇厚,下唇突出,另一個重要的表現是大睾丸症。一些患者還有多動症,攻擊性行為或孤癖症。20%患者有癫痫發作。過去曾認為由于女性有兩條X染色體,因此女性攜帶者不會發病,但由于兩條X染色體中有一條失活,女性雜合子中約1/3可有輕度智力低下。
(3)發病的分子機理:現今在X脆性部們已發現了緻病基因FMR-1,它含有(CGC)n三核甘酸重複序列,後者在正常人約為30拷貝,而在正常男性傳遞者和女性攜帶者增多到150~500bp,稱為小插入,相鄰的Cpg島未被甲基化,這種前突變(premutation)無或隻有輕微症狀。女性攜帶者的CGG區不穩定,在向受累後代傳遞過程中擴增,以緻在男性患者和脆性部位高表達的女性達到1000~3000bp,相鄰的CpG島也被甲基化。這種全突變(full mutation)可關閉相鄰基因的表達,從而出現臨床症狀。由前突變轉化為完全突變隻發生母親向後代傳遞過程中。根據對脆性部位DNA序列的了解,現已可用RFLP連鎖分析、DNA雜交分析、PCR擴增等方法來檢出緻病基因。
(4)治療:Lejeune認為葉酸缺乏是Fra X綜合征時智力低下的原因,他用大劑量葉酸治療患者獲得了良好的效果,但其他作者未能證實葉酸的療效。新近一些作者認為中樞神經興奮劑療效較好,但副作用大。其它有用可樂定(clonidine)、心得安者,據稱可減輕多動症。
3.Y染色體及其結構異常
Y染色體用熒光染色時,因長臂未端有寬闊的極明亮的熒光帶,易與21、22号區别。Y染色體長臂的多态性非常明顯,存在種族差異,大Y在中國人和日本人中的比例較高。
4.Y染色體的數目異常
包括前已述及的XYY以及超Y綜合征(XXYY,X--等)。Y染色體的結構異常包括Y的長臂或短臂缺失、等臂染色體i(Yq)和i(Yp)、環狀染色體和雙着絲粒染色體(為兩條Y的短臂、相連或兩條Y的長臂相融合)、倒位和各種涉及Y的易位(即Y與常染色體,Y與X染色體的易位等)。此外性反轉綜合征46,XX男性是Yq上的SRY基因易位到一條X染色體所緻,而46,XY女性是SRY基因缺失或突變的結果。
單體及部分單體綜合征(常染色體異常)
整條常染色體的丢失通常是緻死的,因而極為罕見,但确有小染色體(如21号)完全丢失的報告。由于易位、環形成或缺失導緻的染色體部分單體則比較多見。現以5q-綜合征,即貓叫綜合征為介紹如下。
貓叫綜合征(5q-綜合征)為最見的缺失綜合征,其發病率估計為1:50000,女性多于男性。患嬰的哭叫聲非常似小貓的咪咪聲,故得名。患兒面部情似很機靈,但實則智力低下非常嚴重(智商常低于20),發育遲滞也很明顯。常見的臨床表現還有小頭、滿月臉、眼裂過寬、内眦贅皮、下颌小且後縮(圖2-20)。約20%患者有先天性心髒病,主要是室間隔缺損和動脈導管未閉等。
患者的染色體缺失片段大小不一。症狀主要由5p15的缺失引起。畸變多數是新發生的。由染色體片段的單純缺失(包括中間缺失)的占80%,不平衡易位引起的占10%,環狀染色體或嵌全體則比較少見,由親代染色體重排導緻的5p-綜合征不多見。
性染色體數目異常綜合征
Klinefelter綜合征
(Klinefelter syndrome)又稱為先天性睾丸發育不全或原發小睾丸症。患者性染色體為XXY,即比正常男性多了一條X染色體,因之本亦常稱為XXY綜合征。
Klinefelter綜合征的發病率相當高,男性新生兒中達到1.2‰。根據白種人的資料,身高180cm的男性患病率為1/260,在精神病患者或刑事收容機構中為1/100,在因不育而就診者中約為1/20。臨床表現為睾丸小而質硬,曲細精管萎縮,呈玻璃樣變。由于無精子産生,故97%患者不育。患者男性第二性征發育差,有女性化表現,如無胡須,體毛少,陰毛分布如女性,陰莖龜頭小等,約25%的患者有乳房發戶。患者身材高。四肢長,一部分患者(約1/4)有智力低下,一些患者還有精神異常及患精神分裂症傾向。實驗室檢查可見雌激素增多,19-黃體酮增高,激素的失調與患者的女性化可能有關。
絕大多數患者的核型為47,XXY。大約有15%患者為兩個或更多細胞系的嵌合體,其中常見的為46,XY/47,XXY;46,XY/48,XXXY。額外的X是由于親代減數分裂時X染色體不分離的結果。
用睾丸酮治療可以收到一定的效果,它可促使第二性征發良并性病患者的心理狀态。
XYY綜合征
在男嬰中的發生率為1:900。XYY男性的表型的正常的,患者身材高大,常超過180cm,偶爾可見隐睾,睾丸發育不全并有精過程障礙和生育力下降,尿道下裂等,但大多數男性可以生育。XYY個體易于興奮,易感到欲望不滿足,厭學,自我克制力差,易産生攻擊性行為。XYY核型是父親精子形成過程中第二次減數分裂時發生Y染色體不分離的結果。
Turner綜合征
又稱為45,X或45,XO綜合征、女性先天性性腺發育不全或先天性卵巢發育不全綜合征。在新生女嬰中的發病率約為0.2‰-0.4‰,但在自發流産胚胎中Turner綜合征的發生率可高達7.5%.患者表型為女性,身材矮小,智力一般正常,但常低于其同胞,面呈三角形,常有睑下垂及内眦贅皮等,上颌突窄,下颌小且後縮,口角下旋呈鲨魚樣嘴,頸部的發際很低,可一直伸延到肩部,約50%患者有蹼頸,即多餘的翼狀皮膚,雙肩徑寬,胸寬平如盾,乳頭和乳腺發育差,兩乳頭距寬,肘外翻在本病十分典型,第四、第五掌骨短而内彎,并常有指甲發育不全。嬰兒期腳背有淋巴樣腫,十分特殊。泌尿生殖系統的異常主要是卵巢發育差(索狀性腺),無濾泡形成,子宮發育不全,常因原發性閉經來就診。由于卵巢功能低下患者的陰毛稀少,無腋毛,外生殖器幼稚。此外,大約有1/2患者有主動脈狹窄和馬蹄腎等畸形。
Turner綜合征的核型除典型的45,X(約占55%)外,還有各種嵌合型和結構異常的核型。最常見的是嵌合型46,XX/45,X和46,X,i(Xq)。一般說來,嵌合型的臨床表現較輕,而有Y染色體的嵌合型可表現出男性化的特征,身材矮小和其它Turner症狀主要是由X短臂單體性決定的,但卵巢發育不全與不育則更多與長臂單體性有關。
Turner綜合征的發病機理是雙親配子形成過程中的不分離,其中約75%的染色體丢失發生在父方,約10%的丢失發生在合子後早期卵裂時。
除少數患者由于嚴重畸形有新生兒期死亡之外,一般均能存活,隻是在青春期才被檢出。其智力發育障礙也較輕,應用激素在14歲以前開始治療可以促進第二性征和生殖器官的發育,月經來潮,心理狀态改變,但不能促進長高,個别患者可生育。
47,XXX和X女性
本病又稱為超雌(superfemale)。發病率約為0.44‰或1/2250。
多數具有三條X染色體的女性無論外形、性功能與生育力都是正常的,隻有少數患者有月經減少、繼發閉經或過早絕經等現象。大約有2/3病智力稍低,并有患精神病傾向。
除了47,XXX外,一些患者的核型為嵌合型,即47,XXX/46,XX。XXX患者的母親生育年齡平均約增高4歲。這表明不分離主要發生在母親一方。少數患者有4條甚至5條X染色體。一般來說,X染色體愈多,智力損害和發育畸形愈嚴重。
性染色體的結構畸變
性染色體的結構畸變
X染色體的結構異常
常見的X染色體結構異常有各種缺失、易位和等臂染色體。它們的臨床表現多樣,主要取決于涉及X染色體上的哪些區段異常,因為不同的區段載有的基因不同,缺失導緻的症狀也不同。Wyss等曾作一Turner綜合征的核型與症狀關系圖。有助于了解X染色體結構異常的表現。
(1)X短臂缺失(XXp-):Xp遠端缺失病人有諸如身材矮小等Turner綜合征特征,但性腺功能正常。Xp缺失如包括整個短臂,則患者既有Turner綜合征的體征,又有性腺發育不全。X染色體長臂等染色體[X,i,(Xq)]的臨床表現與此類似,因為也缺失了整個短臂。
(2)X長臂缺失(XXq-):缺失在q22以遠者,一般僅有性腺發育不全,原發閉經,不育,而無其它諸如身材矮小等Turner綜合征體征。缺失範圍較大,包括長臂近端者,有性腺發育不全外,一些患者還有其客觀存在體征。X染色體等臂染色體[Xi(p)]與此類似。Xq中間缺失累及q13-q26者性腺功能正常,但有其它體征,可見中段缺失與Turner體征出現有關。
通常部分缺失`形成環狀或等臂染色體的X均選擇性地失省事,從而保證有一條正常的X。
(3)易位:當X染色體與常染色體發生平衡易位時,由于基因平衡的保持,一般不會産生症狀,此時失活的正常的X染色體。但如平衡易位斷點在q12-q26時,有活性的X在該區被分為兩部分,就會導緻性腺發育異常。此外,如常染色體節段易位到X染色體産生不平衡易位時,多數産生雙着絲粒染色體,其表型取決于Xp或Xq上斷裂點的位置。
脆性X染色體綜合征
本世紀初一些學者注意到智力下患者中邏輯性多于女性。1943年Martin和Bell在一個家系兩代人中發現11名邏輯性患者和兩名輕度智力低下的女性,認為該家系智力低下是與X連鎖的,因此X連鎖智力低下又稱為Martin-Bell綜合征。
1969年Lubs首先在邏輯性智力低下患者及其女性親屬中發現了長臂具有“随體和呈細絲狀次缢痕”的X染色體。後來,Sortherland證明細絲位位于X染色體長臂2區7帶(Xq27)。它在低葉酸培養條件下表達,并提出了脆性部位(franile site)的概念。現今人們把在Xq27處有脆性部位的X染色體稱為X染色體(fragile X,fra X),而它所導緻的疾病稱為脆性X染色體綜合征。
(1)發病率:本病在邏輯性中的發病率為1/1000~1/1500,僅次于先天愚型。在所有邏輯性智力低下患者約10%~20~為本病所引起。
(2)臨床表現:主要表現為中度到重度的智力低下,其它常見的特征尚有身長和體重超過正常兒,發育快,前額突出,面中部發育不全,下颌大而前突,大耳,高腭弓,唇厚,下唇突出,另一個重要的表現是大睾丸症。一些患者還有多動症,攻擊性行為或孤癖症。20%患者有癫痫發作。過去曾認為由于女性有兩條X染色體,因此女性攜帶者不會發病,但由于兩條X染色體中有一條失活,女性雜合子中約1/3可有輕度智力低下。
(3)發病的分子機理:現今在X脆性部們已發現了緻病基因FMR-1,它含有(CGC)n三核甘酸重複序列,後者在正常人約為30拷貝,而在正常男性傳遞者和女性攜帶者增多到150~500bp,稱為小插入,相鄰的Cpg島未被甲基化,這種前突變(premutation)無或隻有輕微症狀。女性攜帶者的CGG區不穩定,在向受累後代傳遞過程中擴增,以緻在男性患者和脆性部位高表達的女性達到1000~3000bp,相鄰的CpG島也被甲基化。這種全突變(full mutation)可關閉相鄰基因的表達,從而出現臨床症狀。由前突變轉化為完全突變隻發生母親向後代傳遞過程中。根據對脆性部位DNA序列的了解,現已可用RFLP連鎖分析、DNA雜交分析、PCR擴增等方法來檢出緻病基因。
(4)治療:Lejeun認為葉酸缺乏是Fra X綜合征時智力低下的原因,他用大劑量葉酸治療患者獲得了良好的效果,但其他作者未能證實葉酸的療效。新近一些作者認為中樞神經興奮劑療效較好,但副作用大。其它有用可樂定(clonidine)、心得安者,據稱可減輕多動症。
Y染色體及其結構異常
Y染色體用熒光染色時,因長臂未端有寬闊的極明亮的熒光帶,易與21、22号區别。Y染色體長臂的多态性非常明顯,存在種族差異,大Y在中國人和日本人中的比例較高。
Y染色體的數目異常
包括前已述及的XYY以及超Y綜合征(XXYY等)。Y染色體的結構異常包括Y的長臂或短臂缺失、等臂染色體i(Yq)和i(Yp)、環狀染色體和雙着絲粒染色體(為兩條Y的短臂、相連或兩條Y的長臂相融合)、倒位和各種涉及Y的易位(即Y與常染色體,Y與X染色體的易位等)。此外性反轉綜合征46,XX男性是Yq上的SRY基因易位到一條X染色體所緻,而46,XY女性是SRY基因缺失或突變的結果。