概述
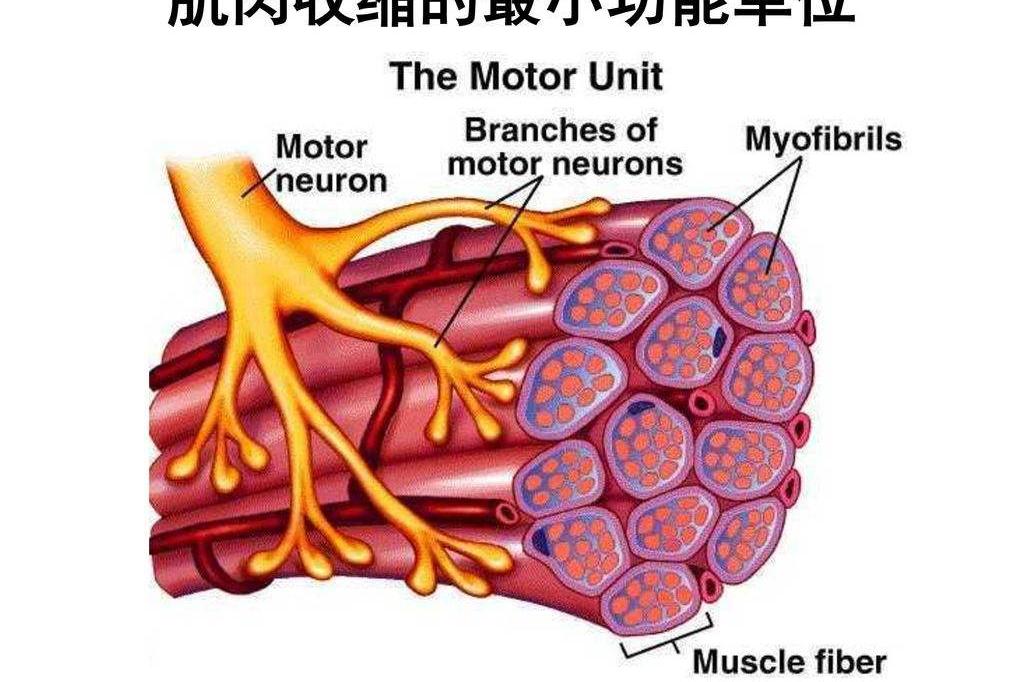
脊髓運動神經元發出的運動神經纖維通過終闆支配骨骼肌的運動。一個運動神經元和它所支配的全部骨骼肌纖維所組成的結構和機能單位叫做一個運動單位。運動單位的生理特點是作為一個整體活動。
每一個運動神經元支配的肌纖維數目不同,它的軸突可以和100~160條的肌纖維發生突觸聯系。貓的腓腸肌包括約430個運動單位。有的一個運動神經元支配較多的肌肉纖維,如四肢肌群,因而其運動粗笨有力;有的一個運動神經元僅支配很少,甚至一個肌細胞,如眼肌,因此運動十分精細。
運動單位病分類
運動單位病常見于兒童,病種繁多,可分為肌源性與神經源性、遺傳性與非遺傳性、先天性與後天獲得性、急性與慢性、進行性與靜止性。許多疾病的臨床特征相互重疊交錯,表現為遺傳性及肌無力等進行性加重的特點。診斷與鑒别診斷,主要靠實驗室檢查,如谷草轉氨酶(GOT)、乳酸脫氫酶(LDH)、肌酸磷酸激酶(CPK)等酶學檢查,以及肌電圖(EMG)、肌活檢、頭顱CT與MRI等神經電生理與影像學檢查。所謂肌源性肌萎縮是指由肌纖維病變導緻的肌萎縮。神經源性肌萎縮,指脊髓前角細胞、運動神經纖維、神經肌肉接頭的病變産生的肌萎縮。
臨床常見的運動單位病
脊髓前角細胞變性——脊髓性肌萎縮
脊髓性肌萎縮(spinalmuscularatrophy,SMA)是常染色體隐性遺傳病,本病特點是脊髓前角細胞變性。臨床表現為進行性、對稱性肢體近端為主的廣泛性遲緩性麻痹與肌萎縮,智力發育及感覺發育正常。其發病與運動神經元存活基因(SurvivalMotorNeuron,SMN)端粒側exon7的純合性缺失或突變有關,重型SAM主要與神經元凋亡抑制蛋白基因(neuronalap-optosisinhibitoryprotein,NAIP)有一定關系。病理特點為脊髓前角細胞變性和減少。臨床上分為嬰兒型、少年型及中間型三類。嬰兒脊髓性肌萎縮(Ⅰ型)又稱Werding-Hoffmann病。6個月以前發病,存活率低。中間型脊髓性肌萎縮(Ⅱ型)又稱慢性嬰兒型,生後18個月以内發病,進展緩慢。少年型脊髓性肌萎縮(Ⅲ型)即慢性脊髓肌萎縮,又稱Kagelberg-Weland-er病。2歲以後發病,進展緩慢。
神經肌肉接頭病——重症肌無力
重症肌無力(myastheniagravis,MC)是神經肌肉接頭處的傳遞障礙所緻的自身免疫受體病。臨床特點是自主運動時肌肉有明顯疲勞性和無力表現,經休息或用膽堿酯酶抑制劑治療後症狀減輕或消失。
神經肌肉接頭(neuromuscularjunction,NMJ)是由神經終末(突觸前)、肌膜的中闆(突觸後膜)和間隙(突觸間隙)所組成,使沖動從運動神經末梢迅速、有效地傳遞至骨骼肌纖維。臨床上本病有不同類型,以後天獲得性肌無力在小兒稱為少年型重症肌無力最常見。
少年型重症肌無力(juvenilemyastheniagravis,JMG)是累及神經肌肉接頭處突觸後膜AchR的自身免疫病。病因不明,免疫學異常的病因尚無定論,遺傳可能為内因,而在外因中多數學者認為與胸腺的慢性病毒感染有關。病理表現為肌纖維呈現非特異性炎症改變,神經接頭處突觸後膜有免疫複合物沉積,胸腺增生或淋巴結生發中心增生。臨床起病多在2歲以後,女多于男,臨床分為眼肌型、全身型即腦幹型。
肌病——進行性肌營養不良
進行性肌營養不良(progressivemusculardystrophy,PMD)是一組遺傳性肌肉變性病,其臨床主要特征是進行性肌萎縮與無力,臨床幾種類型中假性肥大型營養不良(Duchennemus-culardystrophy,DMD)又稱Duchenne型肌營養不良最常見。該型是X連鎖隐性遺傳,男性發病,女性攜帶基因。發病主要為基因缺少(Xp21.1~21.3),現已闡明DMD基因的表達産物是肌營養不良蛋白(dystrophin,Dp),病理改變為肌纖維大小不等,肌纖維變性、壞死與再生并存。肌纖維肥大的部分呈玻璃樣變,其間有大量脂肪和結締組織即假性肥大。臨床起病多在3~5歲,表現為運動發育遲緩。
代謝性肌病——線粒體疾病和線粒體腦肌病
線粒體肌病(mitochondrialmyopathic)和線粒體腦肌病(mitochondrialencephalomyopathy)是一組由線粒體結構和功能異常所緻的疾病,以骨骼肌受累為主稱線粒體肌病,如同時累及中樞神經系統則稱為線粒體腦肌病。線粒體是細胞内除細胞核以外隻有這一個DNA複制系統的微器官,其主要功能是合成三磷腺苷(ATP),為細胞代謝提供足夠的能量。認為線粒體基因缺陷是導緻本組疾病的主要原因。由于遺傳基因缺陷造成線粒體代謝過程中酶缺失或活性降低,導緻線粒體代謝過程中所需要的脂肪酸、糖原等不能被線粒體有效的利用,因此産生氧化代謝的問題,最終不能給細胞提供足夠的能量。線粒體腦肌病臨床表現為以下6種類型:
(1)Kearns-Sayre綜合征(KSS),主要表現為眼外肌癱瘓,視網膜色素變性及心髒傳導阻滞三聯症。
(2)慢性進行性眼外肌癱瘓(chronicprogressiveexternalophthalmoplegia,CPEO),主要表現為進行性眼外肌癱瘓,包括上睑下垂、眼球活動受限。
(3)線粒體腦肌病-乳酸酸中毒-卒中樣發作綜合征(mitochondrialencephalomyopathywithlacticacidemiaandstroke,MELAS),10歲左右發病,特點為卒中樣發作,血中乳酸、丙酮酸增高,局限性或全身性癫痫發作。
(4)肌陣攣性癫痫伴破碎紅纖維(Myoclonusepilepsywithragged-red-fiber,MERRF),主要表現為肌陣攣發作,可伴小腦共濟失調。
(5)Leigh綜合征,又稱壓急性壞死性腦脊髓炎(subacutenecrotizingencephalomyelopathy,SNE),臨床主要表現腦脊髓受累,如精神運動遲滞、運動障礙、進行性淡漠等。
(6)Alper綜合征,為複合體Ⅳ缺陷所緻,臨床表現為發育遲緩及退化、肌陣攣發作、共濟失調、痙攣性截癱等。
(7)線粒體腦心肌病,臨床特征為全身乏力,X線表現為重度心肌肥大。
周圍神經病——格林巴利綜合征
格林巴利綜合征(Guillain-barre綜合征,GBS)是一種小兒常見的由體液免疫、細胞免疫及細胞因子介導的自身免疫性周圍神經病。臨床分型為急性炎性脫髓鞘多神經病(AIDP)與慢性炎性脫髓鞘多神經病(CIDP),其特征為急性對稱性弛緩性麻痹(包括四肢、顱神經及呼吸肌)、感覺障礙、自主神經功能障礙、腱反射消失。康複主要是改善肌力,預防肌萎縮和關節攣縮變形,促進肢體功能恢複。